Hammer Group
1.1 Structural optimization
The Hammer group is working on developing methods that can perform global optimization of atomistic structure.
Textbook example
Here is an example of a Monte Carlo optimization of 12 Lennard-Jones atoms trapped in two dimensions:
Figure 1: Example from 1st year course at Aarhus University.
Real materials
For real materials global optimization of the atomistic structure can be done using density functional theory (DFT) which is based on a quantum mechanical descriptions of the electrons in the electrostatic potential of the atoms. Here is an example where a favorable reconstructed step structure on rutile-TiO(110) was identified via global optimization (using an evolutionary algorithm) and DFT:

Figure 2: Left and top right: the unreconstructed step structure. Right bottom: The reconstructed step, where the purple Ti atom takes a position which is not an extension of the bulk crystal structure. From: Phys. Rev. B 84, 205434 (2011).
1.2 DFT and ML
The computational cost
Whenever DFT is employed, a very realistic description of material properties can be obtained, but at the expense of the computation requiring a lot of computational resources.
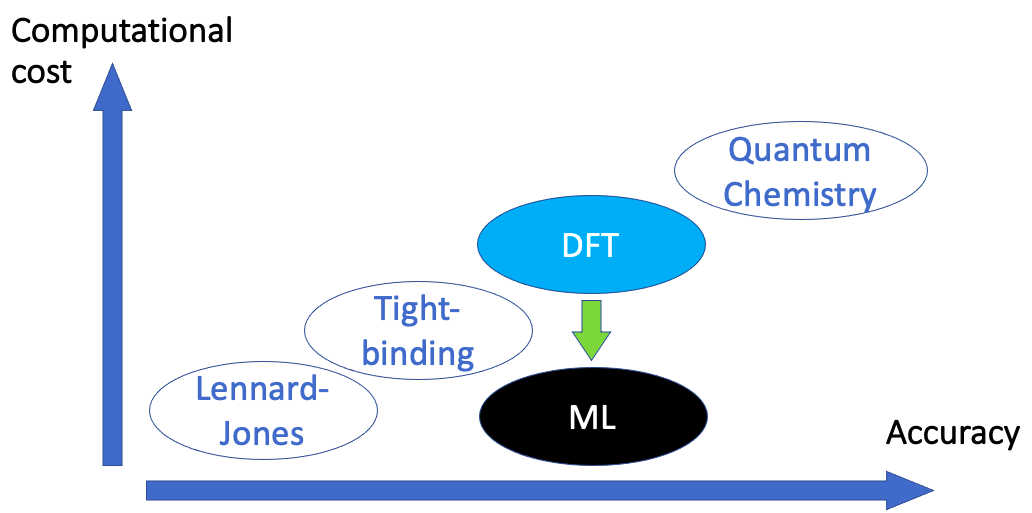
Figure 3: Illustration of the trade off between accuracy and computational cost. If machine learning (ML) methods can be introduced, the DFT level of accuracy may be retained while the computational cost is reduced.
Introducing machine learning methods in density functional theory (DFT) calculations serves to bypass some of the costly DFT calculations based on the previously done expensive DFT calculations. Consider that some structures are described by coordinates, , and that it is known from DFT calculations that these structures have energies . Then a useful machine learning method would be one that given those coordinates reproduce the energies and at the same time predict reasonable energies, , when presented with other structures described by coordinates, . This is illustrated here:
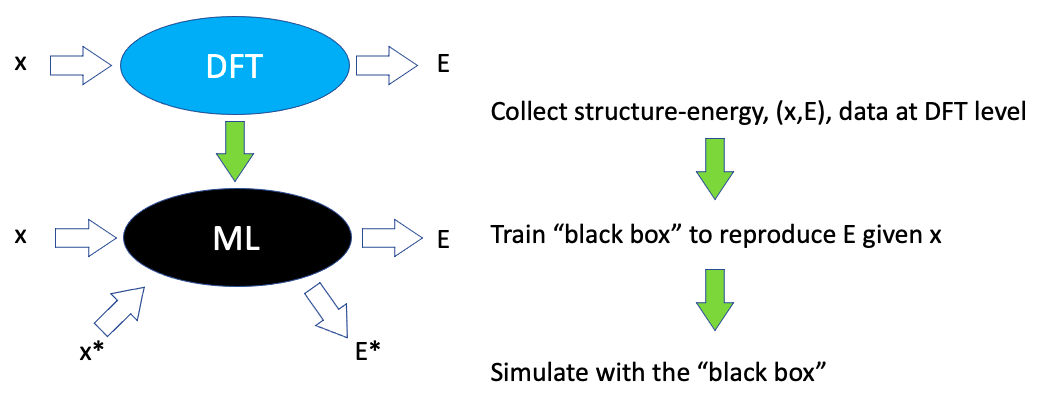
Figure 4: DFT: The expensive DFT program provides energies when given coordinates . ML: The inexpensive machine learning program does the same and gives reasonable energy estimates, , for other structures, .
1.3 Computer code
Our code is called Atomistic Global Optimization X (AGOX) where X refers to the various different methods (basin hopping, evolutionary algorithm, GOFEE, etc) that have been implemented.
The code can be found on gitlab here and the documentation is found here. A paper is found in J Chem Phys or in its preprint version on arXiv.
1.4 ML Methods
Some recent methods developed:
2022: SOAP-based Gaussian Process Regression (GPR) for the ML-potential
2020: GOFEE - combining a GPR method with an evolutionary search method. The search is done in the lower confidence bound,
where is the error estimate and .2018: LEA - combining a neural network for energy and force prediction with an evolutional search method.